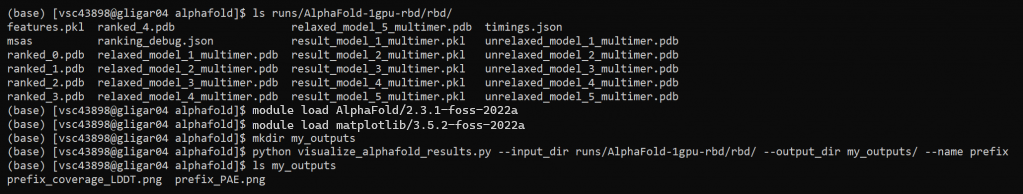
Basic workflow example (ssh)
A full instruction set for running a new protein through AlphaFold is described in this section.
1. Log in using your credentials (details)
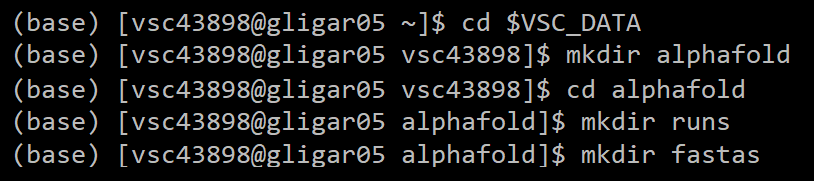
2a. If running for the first time: set up directories
After this, copy the content of the job script to the nano program on the command line (details on how to work with nano), or copy it from your personal computer using WinSCP. Note that pasting in PuTTY is done with a right-click by default.

2b. If directories already set up: navigate to the alphafold/ directory
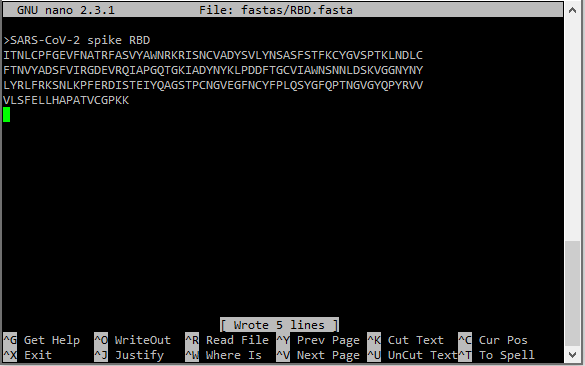
3. Create a new FASTA file

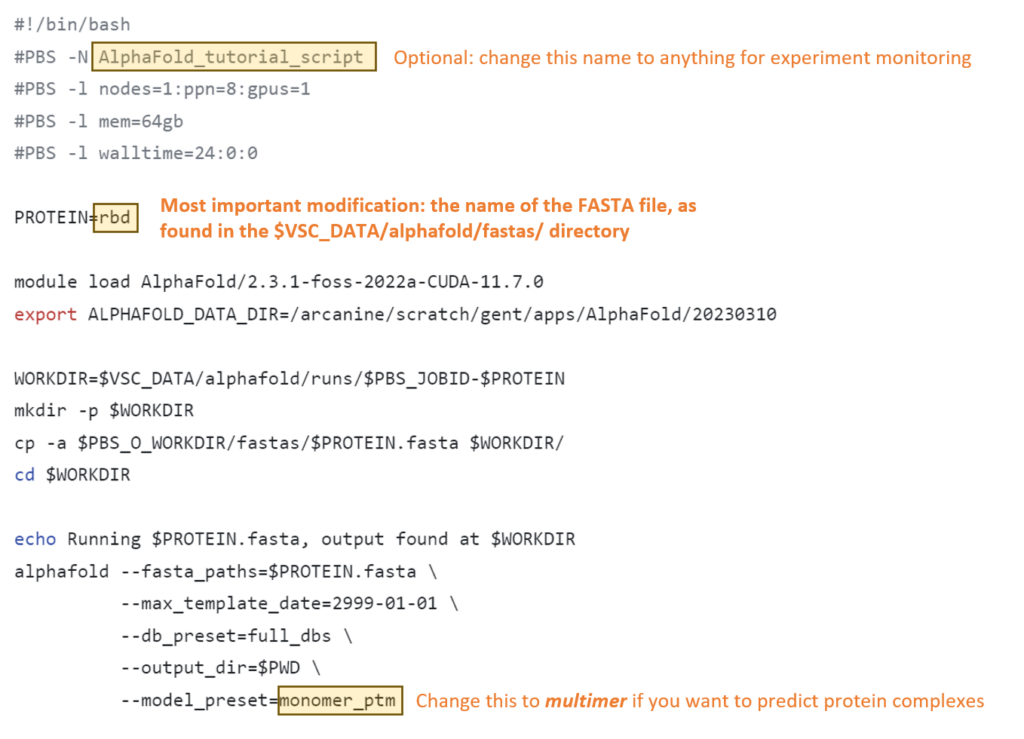
4. Modify the job script to use your FASTA file
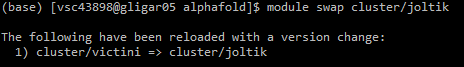
5. Swap to the GPU cluster
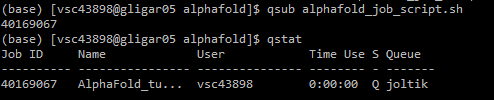
Q) will change to R when running, F when failed, and C when completed (or crashed). 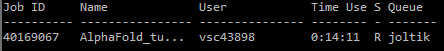
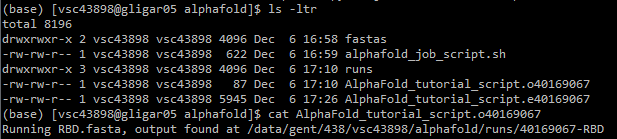
alphafold/. 7. Gather outputs
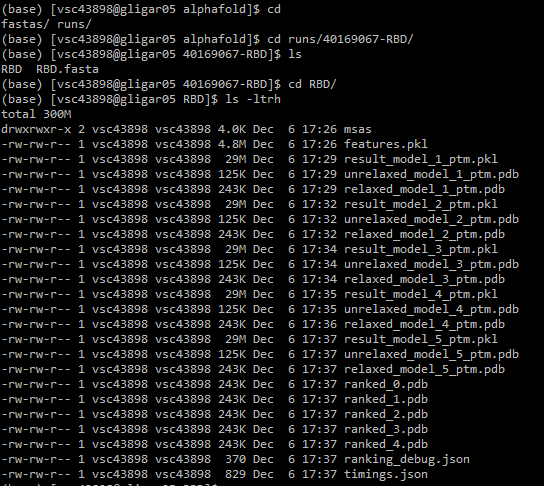
runs/. Copy the results you need (mainly the ranked_0 to 5 .pdb files to your personal computer, using for instance WinSCP). 8. Generate visualizations
As described in an earlier topic, you can generate visualizations for this run using an extra python script. Load in the correct modules (AlphaFold and matplotlib) and run visualize_alphafold_results.py as shown below.