Computational limits
The HPC hosts powerful GPU clusters, essential to run any deep learning experiment of significant size. Depending on the cluster available to you, there is a limited amount of GPU memory and CPU memory (RAM). Larger GPUs are necessary for longer sequences, while RAM can be a limiting factor if MSAs grow too large.
Extra: AlphaFold documentation (VUB, Hydra) can be found at: https://hpc.vub.be/docs/software/usecases/#alphafold
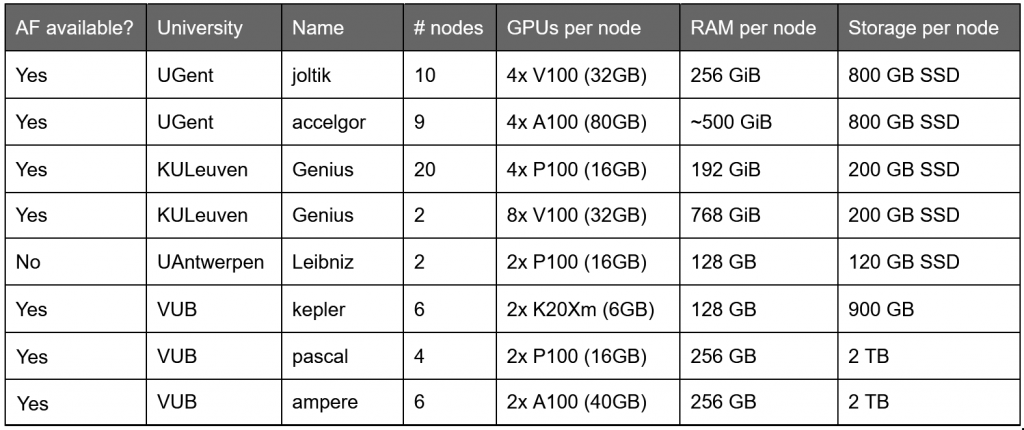
To give an estimate of the requirements to run AlphaFold, a series of experiments were run on the UGent joltik and accelgor clusters. Here, the required RAM with in function of input sequence length was measured, as well as the time elapsed for the different stages in prediction: the MSA search, the prediction models, and the relaxation of the resulting 3-D structures. Twenty-two sequences were arbitrarily chosen, with sequence lengths ranging from 200 to 3000 amino acids. The experiments were done with the default MSA search setup, with AlphaFold version 2.0.0.
This graph suggests that the maximum memory usage is not necessarily directly linked to primary sequence length, but rather to the MSA size. Short sequences can already surpass the required memory (and make the experiment crash), while long sequences might perfectly pass.
Looking at GPU limits, it is possible to run sequences of up to approximately 1000 residues on any 12 GB GPU, such as the ones that are sometimes allocated on Google Colab, of up to 1400 residues on 16 GB GPUs that are available on KULeuven, VUB and Google Colab systems. Larger GPUs, such as the 32GB, 40GB and 80GB units at the KULeuven/UGent/VUB clusters, can host sequences of almost 3000 residues and more.
Finally, one sequence with length ~2300 residues, depicted in red, made the system crash due to an inherent allocation limit of the libraries used in AlphaFold. Though not solvable at present, this error should only very rarely occur.
To give an idea of required computation time (not including HPC job queues), the different stages of the AlphaFold predictions were also timed for the previous experiments. Note that the following conclusions are based on results on the joltik cluster, and do not necessarily transfer to other clusters.
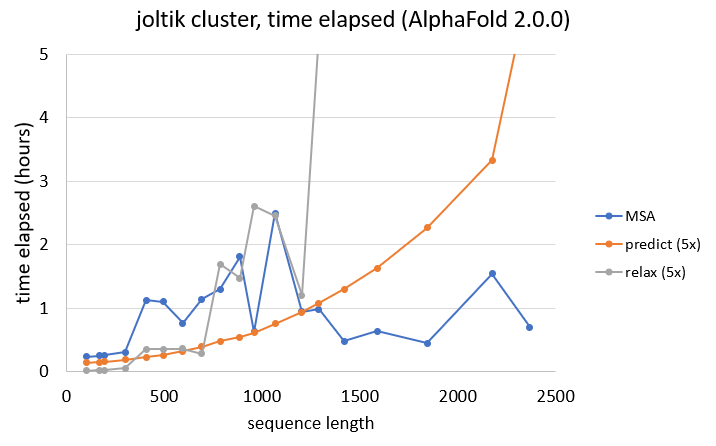
As also seen for the memory requirements of the MSA search, the time elapsed does not necessarily solely depend on sequence length. Longer sequences do not necessarily require more time than shorter ones.
When looking at prediction times, summed for all five AlphaFold models, we can see that the time elapsed increases quadratically. In general, for sequences with less than ~1200 residues, more time is spent on the MSA search than on the actual predictions.
Finally, AMBER relaxation is even more dependent on sequence length, with sequences above 1000 residues taking a very long time to relax all five predicted structures.
As a final remark, note that it is not impossible to run AlphaFold on non-GPU systems. However, a significant increase in computation time is to be expected. The increase in elapsed time is situational, but as an example, one of the above sequences, with 301 residues, used just eight minutes for the prediction part on the GPU, but more than four hours on the CPU of the skitty cluster (note that the usage of the CPU cluster was not optimized and that an improvement in performance might still be possible, though the magnitude difference would remain). Another sequence, with 787 residues, used half an hour on the GPU, but ran for more than 17 hours on the CPU of the skitty cluster.