Prepare directories and files
The following steps are necessary to prepare for an AlphaFold run:
- Set up directories
- Create a FASTA file
- Set up job script
- Swap to GPU cluster
1.
The job script, the inputs to AlphaFold, as well as its outputs, will all need to be stored somewhere in your directories on the HPC. As a user, you get a limited space quota allocated to your account. Information on the used and remaining storage capacity can be found on the VSC user portal (https://account.vscentrum.be/). To summarize, by default you have two file systems: $VSC_HOME (~6GB) and $VSC_DATA (~24GB). We recommend to make use of the $VSC_DATA file system, as AlphaFold potentially generates large amounts of data, ranging from several MBs up to several GBs. If more frequent usage is desired, more storage will probably be required. We recommend to look into setting up a virtual organization (VO) for your research group, or to join an existing one. Multiple TB of storage can be made available in this way. The data directory to use in this case (instead of $VSC_DATA) would be $VSC_DATA_VO_USER. This is the location for your own VSC account within your VO.
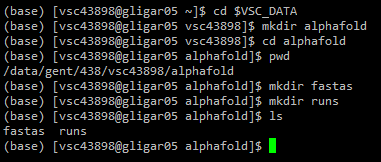
Within the chosen directory, make a directory called alphafold. Everything related to running AlphaFold will be located here. In this directory, create two new directories: fastas and runs. In fastas, the input FASTA files will be stored. In runs, all outputs generated will be automatically stored.
2.
To create a FASTA file, either copy a file from your personal computer to the HPC using the SCP protocol, or create a new FASTA file on the command line and copy the contents there.

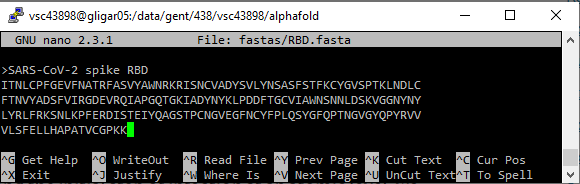
nano to create or edit text files. Pasting content from the clipboard can be achieved by right clicking the terminal window. To save a file in nano, press Ctrl+O and Enter. To exit, press Ctrl+X. If the file is changed but not yet save, nano will prompt you; insert Y and press Enter to save modifications, insert N and press Enter to discard them.The FASTA file should contain either a single protein sequence (for monomer folding), or multiple sequences when folding protein complexes with AlphaFold-Multimer.
3.
The job script can be edited using the nano command. For more details on how to modify this file to your liking, see the next lesson.
4.
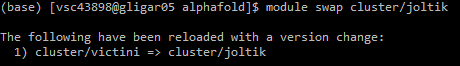
Before you can submit the job script, you need to swap to the correct cluster. As AlphaFold runs on GPU, options here are limited. For example, for Ghent University users the joltik cluster is available. Swap to the appropriate cluster using the module swap cluster/joltik command: