Setting up the job script
Note that the scripts described here were developed for the AlphaFold installation on the UGent HPC; some directories, environment variables and modules are different on other systems.
Before running AlphaFold, first make sure that you have the right files and directories set up, and that you have loaded the correct GPU module.
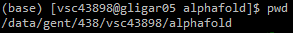
alphafold subfolder.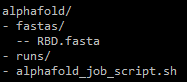
alphafold/, you should have a fastas directory with one or more FASTA files for which you want to fold the protein(s).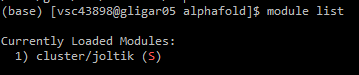
module list or ml, verify whether the desired cluster is selected.Below you can find the contents of the alphafold_job_script.sh file. You can edit it with the nano command, for example. You can download the file contents here. Once everything is configured, you can submit the script by running qsub alphafold_job_script.sh.
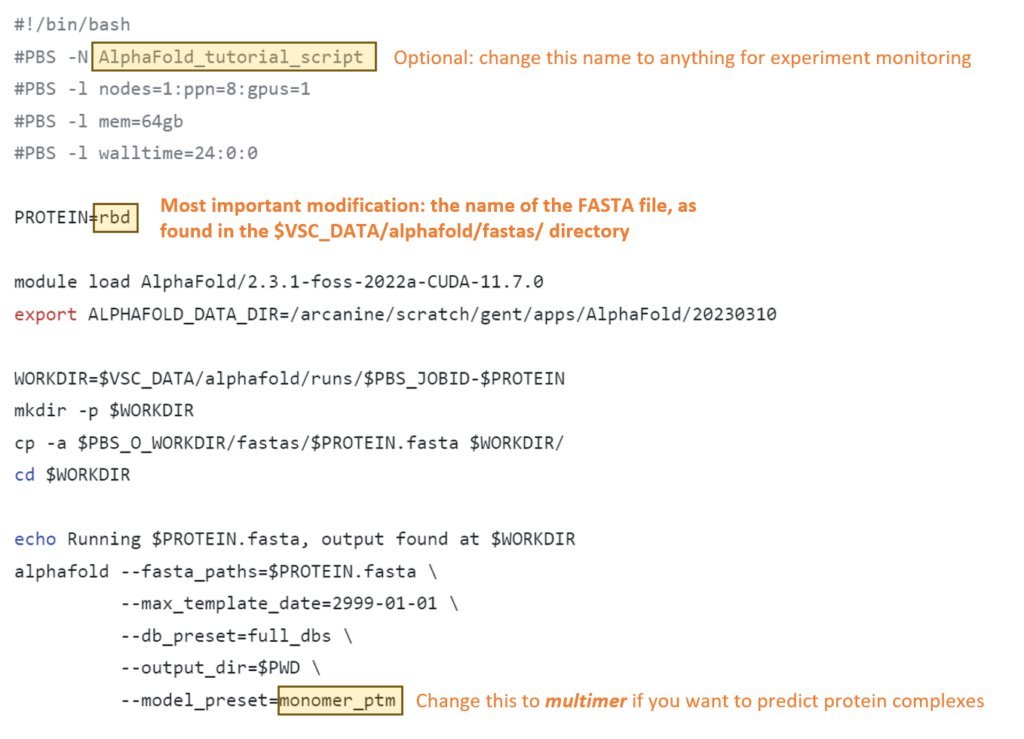
For regular use, you need to change just one line in the script. Additional lines to change could be the name of the experiment (could be useful for experiment monitoring but is not strictly necessary), and the usage of the multimer model.
This code might look scary and difficult at first glance, but it can remain largely unchanged for running multiple experiments. Below you can find an overview of the few things you would ever want to change.
Job script details:

Changes to make for individual experiments:
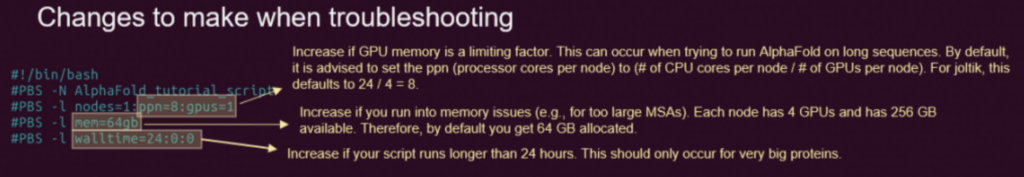
When running large proteins, you might run into problems with the default settings. These are the parameters that you might want to change, based on feedback that you can find on the troubleshooting page.