Overview of the architecture
Developed at DeepMind, the AlphaFold architecture is a delicate collaboration between different modules, trained using end-to-end learning. This means that all parameters in the network are trained at once, from input to output, without the need of independently finetuning individual modules.
The architecture is depicted as follows. Note that the visualizations below were copied and adapted from the official AlphaFold paper (Jumper et al, 2021).
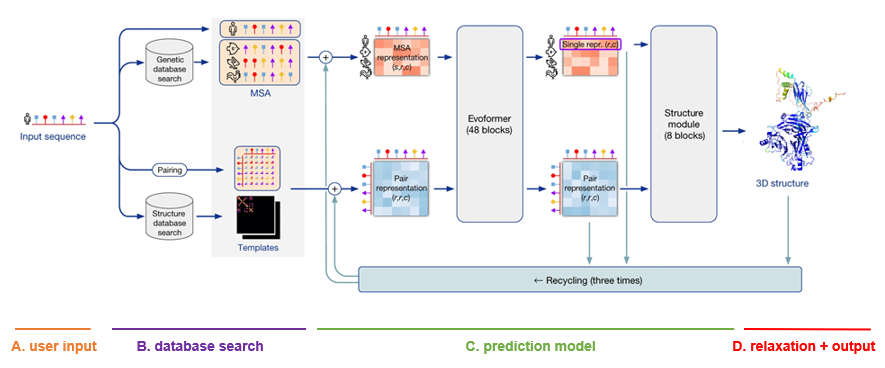
A. User input
The only input that the user needs to supply to AlphaFold in order to make a prediction, is a FASTA file with the protein primary sequence of interest. In the case of AlphaFold-Multimer, multiple proteins can be specified within the FASTA file.
B. Database search
For each sequence specified, a multiple sequence alignment (MSA) search is launched. This is done by first running JackHMMER on Mgnify (keeping the top 5’000 matches) and UniRef90 (keeping the top 10’000 matches), and then running HHBlits on UniClust30 + BFD (keeping all matches).
As AlphaFold bases itself largely on coevolutionary information, a qualitative and deep MSA is essential for good predictions. In their publication, DeepMind claims that they see a significant drop in accuracy for MSAs of less than 30 sequences. In case of protein complexes, MSAs are paired according to evolutionary distance (prokaryotes) or simply the ranking of the matches (eukaryotes).
Finally, although less important than MSA depth, a template search is done. Using the MSA obtained from UniRef90, PDB70 is searched with HHSearch, only allowing templates before a specified date to be found. After discarding templates identical to (a subset of) the input sequence, the top 4 templates are chosen. These templates serve as a starting position for the prediction models.
C. Prediction model
The MSA and templates are given to five AlphaFold models. These all have the same network architecture, but different parameters following five independent training stages with different randomization seeds. Thus, they will predict slightly different 3-D structures.
The network architecture has two main parts. It consists of Evoformer blocks, which apply pairwise updates to numerical MSA representation and a 2-D pair representation, and Structure module blocks, which take care of the actual folding. These modules are repeated multiple times via a process called recycling, where the predicted 3-D structure is used as an input template for a new prediction iteration for further finetuning. By default, three recycling runs are done.
D. Relaxation + output
After each model predicts a 3-D structure, AMBER relaxation is done. Structures are ranked by the average predicted local distance difference test (pLDDT), found in the output of the prediction models. The pLDDT can be seen as a measure of local prediction confidence per position.
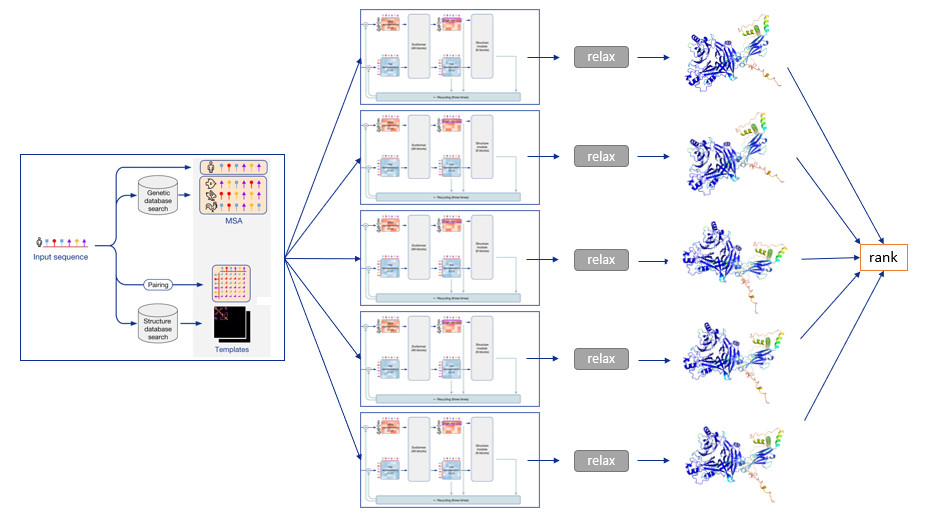
To summarize, the following diagram illustrates the full prediction process: one database search is done to find MSAs and templates, and the exact same input is given to five identical neural network architectures, though parameterized differently. This yields five 3-D structures with tiny or big differences, which are optionally relaxed and finally ranked according to the model’s confidence.
References:
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2